Polycythemia Vera
Introduction
Polycythemia vera (PV) is a clonal stem cell disorder characterized by erythrocytosis, often accompanied by leukocytosis and/or thrombocytosis. The disease is associated with burdensome symptoms, reduced quality of life, risk of thrombohemorrhagic complications and risk of transformation to myelofibrosis (MF) and acute myeloid leukemia (AML).1
About the disease
Erythrocytosis is the most prominent clinical feature of polycythemia vera (PV) and distinguishes it from other myeloproliferative neoplasms (MPNs). Similar to other MPNs, individuals with PV often have splenomegaly and significant burden of disease-related symptoms, including pruritus, night sweats, fatigue, and bone pain.2
Patients with PV are also at risk of thrombotic complications and transformation to secondary myelofibrosis also known as post-polycythemia vera myelofibrosis (PPV-MF) or acute myeloid leukemia (AML).3
Thrombosis is one of the major causes of death among PV patients.
Overactive JAK signalling caused by the JAK2 V617F mutation within exon 14 (present in ~ 95% of PV patients) and by different mutations within exon 12 of the JAK2 gene (present in about 4% of PV patients) has been implicated in the pathogenesis of PV.4
The clinical presentation of PV usually involves the following three common scenarios:
- An incidental discovery of elevated hemoglobin or hematocrit
- Diagnosis after a thrombotic event
- Diagnosis after investigating disease-related symptoms5
Natural history
PV patients are at risk of micro- and macrovascular complications, often experience constitutional symptoms, and are at risk of disease progression and associated long-term complications.6
Approximately 10% of PV patients transform into post-polycythemia vera myelofibrosis (PPV-MF), with progressive splenomegaly, MF-related symptoms, and anemia.7
Evolution to acute myeloid leukemia (AML) is rare, with the risk of leukemic transformation between 2% and 7%. Predictive factors are not well known.8
Patients with PV have a shortened life expectancy compared with the age and sex-matched general population. Compared with the general population, the 10-year survival is about 28% lower in PV patients.9
About 45% of PV-related deaths are associated with cardiovascular disease.10
Epidemiology
The estimated incidence of PV worldwide is about 0.84 per 100,000. Recent data from two large health plans in the United States indicate prevalence rates of 44 to 57 cases per 100,000. However, there is wide variation in both prevalence and incidence estimates across data sources.11
The median age at presentation is in the sixth decade and approximately 10% of patients are under 40 years, with an equitable gender distribution.12
The exact cause of polycythemia vera is unknown.
Signs and symptoms
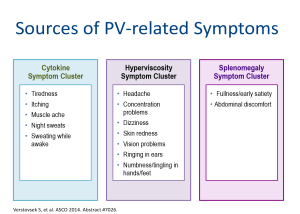
Symptoms experienced by PV patients can be divided into those related to:
- Cytokine production
- Hyperviscosity-related symptoms
- Splenomegaly-related symptoms13
Some disease-related symptoms seen in PV are nonspecific complaints, such as headache, weakness, dizziness, and excessive sweating, which are present in 30% to 50% of PV patients.14
Symptoms more specific to PV include:
- Pruritus, especially after warm baths or showers (aquagenic pruritus; reported by 70% of patients)15
- Erythromelalgia, or a burning pain in the feet or hands accompanied by erythema (seen in 28% of patients).16
Symptom frequency and severity in PV is similar to those experienced by patients with myelofibrosis.17
Compared with the general population, patients with PV report significant increase in fatigue and insomnia.18
Increase in fatigue may be related to iron deficiencies often found in PV patients.19
Diagnosis
When to suspect PV
PV is often first suspected because of an abnormal complete blood count (CBC; e.g., Hb > 185 g/L in men or > 165 g/L in women), but it must be considered in patients with suggestive symptoms.
PV usually affects men and women in their 60s and is rarely seen in people under 40.
- Occasionally, increased red cell volume and viscosity cause weakness, headache, light-headedness, visual disturbances, fatigue, and dyspnea.
- Pruritus often occurs, particularly after a hot bath.
- The face may be red and the retinal veins engorged.
- The palms and feet may be red, warm, and painful, sometimes with digital ischemia (erythromelalgia).
- Hepatomegaly is common, and about 75% of patients have splenomegaly.
- Thrombosis may cause symptoms in the affected site (leg pain, swelling or both with lower extremity thrombosis, unilateral vision loss with retinal vascular occlusion).
- Low-grade fevers and weight loss caused by hypermetabolism.
Diagnostic criteria

Currently the diagnosis of polycythemia vera (PV) is based on the 2008 World Health Organization (WHO) criteria and requires the composite assessment of clinical and laboratory features.20 The 2008 WHO criteria are, however, undergoing revisions.21
The rationale for the proposed changes is based on recent observations that some JAK2 V617F-positive PV patients present with hemoglobin levels lower than the current WHO threshold. Compared with overt PV patients, masked PV (mPV) patients have increased risk of thrombosis, perhaps resulting from late diagnosis and inadequate disease control.22
Basic investigations
Complete blood count (CBC)
For diagnostic purposes, a CBC is of particular relevance, as an increase in all three lineages (erythrocytosis with leukocytosis and/or thrombocytosis) is more indicative of PV than isolated erythrocytosis.23 In patients with isolated erythrocytosis, causes of secondary polycythemia should be considered.
JAK2 V617F mutation
The investigations for PV in suspected cases begin with peripheral blood screening for JAK2 V617F mutation.24
Serum erythropoietin
The possibility of a false positive or false negative mutation test result can be addressed by the concomitant testing of serum erythropoietin (EPO) level, as more than 85% of patients with PV have low serum EPO concentrations. EPO levels above normal are unusual for PV and suggest secondary erythrocytosis, with a specificity of 98%.25
JAK2 exon 12 mutation
Low serum EPO levels in the absence of JAK2 V617F require additional mutational analysis for JAK2 exon 12 mutations.26
Supportive investigations
Bone marrow
Bone marrow (BM) evaluation in JAK2 V617F-positive patients with erythrocytosis provides limited additional value for diagnostic purposes and currently is not routinely performed in Canada.
However, a baseline BM biopsy might be essential in cases where the diagnosis is unclear.
Information regarding age-adjusted BM cellularity and grade of fibrosis may have prognostic value and, as such, help in optimizing therapeutic approaches.27
Cytogenetics
Cytogenetic studies are not routinely performed in PV patients in Canada.
Approximately 11% of PV patients have cytogenetic abnormalities, including trisomy 8, trisomy 9, 13q-, and 20q-.28
These abnormalities are not specific to PV, are more common in older PV patients (>60 years of age), and increase in frequency with disease progression and transformation.29
Earlier studies suggested that some cytogenetic abnormalities may have prognostic value.30
Diagnostic algorithm for PV
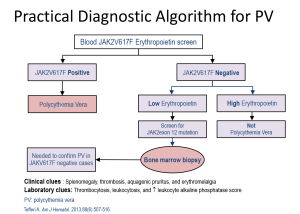
Download PDF
Investigations for PV should begin with screening for JAK2 V617F mutation and testing of serum erythropoietin levels. This should be followed by mutational analysis for JAK2 exon 12 mutations in V617F negative patients with subnormal serum erythropoietin levels. Currently, routine bone marrow evaluation in JAK2 V617F-positive patients is not required for diagnostic purpose, however BM may provide prognostic information and should be considered in all young patients.31
Prognostic risk factors
Polycythemia vera (PV) is associated with a shortened life expectancy compared with the general population.32
Risk factors in PV can be divided into 4 categories including those contributing to:
- Higher incidence of thrombosis
- Reduced survival
- Increased transformation to post-polycythemia vera myelofibrosis (PPV-MF)
- Increased transformation to acute myeloid leukemia (AML)
Risk factors for thrombosis
Thrombosis is much more common in PV than in the general population and it is one of the major causes of death among PV patients.33
At PV diagnosis, arterial thrombosis is present in 16% to 27% and venous thrombosis in 7% to 12% of patients, and the rate of thrombosis is 2% to 4% per year over the course of the disease.34
Thus, risk-stratification of PV patients is typically based on the likelihood of thrombotic risk rather than survival or risk of transformation to PPV-MF or AML.35
Age and history of thrombosis are the two main risk factors associated with thrombosis.36
Risk factors for reduced survival
Advanced age, leukocytosis, history of thrombosis, and abnormal karyotype have negative impact on survival in PV patents.37
Risk factors for transformation to PPV-MF

About 10% of PV patients transform into post-PV myelofibrosis.
Approximately 10% of PV patients transform into post-PV myelofibrosis (PPV-MF), with progressive splenomegaly, myelofibrosis-related symptoms, and anemia.38
Evolution to myelofibrosis is difficult to foresee, although disease duration (>10 years) and allele burden (>50%) are associated with a higher risk of evolution to PPV-MF.39
Risk factors for transformation to AML
Evolution to acute myeloid leukemia (AML) is rare, and predictive factors are not well known.
The risk of leukemic transformation has been reported at 2.3% at 10 years and 5.5% at 15 years, with older age, abnormal karyotype, and leukocytes ≥15 X 109/L as independent risk factors.40
Post PV AML is an aggressive disease with very poor outcomes. Intensive chemotherapy has a limited role in management unless further consolidated by allogeneic transplant. Hypomethylating agents and/or experimental therapies should be considered.41
Assessment Tools
Assessment tools can be used to assess patient risk of cardiovascular event, symptom burden or disease progression.
Cardiovascular risk assessment
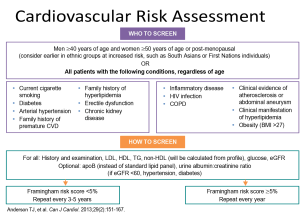
Currently, there are no data on lipid or blood pressure target ranges for individuals with PV. However, with the increased risk of arterial thrombosis, it is prudent to manage atherosclerotic risk factors (including hypertension, hyperlipidemia, and diabetes) and encourage smoking cessation. Clinicians should refer to the Framingham Heart Study and the risk assessment tool incorporated into Canadian Cardiovascular Society guidelines for general prevention of cardiovascular disease.42
Symptom assessment
PV-related symptoms are troublesome to patients, and alleviation of this burden represents a paramount treatment objective. As symptoms develop gradually over time, some patients attribute some of them (i.e., fatigue and loss of functionality) to advanced age.43
It is therefore important for physicians carefully assess patients’ symptoms.
IPVS stratification
Based on age, leukocyte count and history of thrombosis, the International Polycythemia Vera Study (IPVS) identified 3 distinct risk groups.45
The IPVS risk calculator is available online.
Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF TSS)
A recent cluster analysis suggested that symptom burden in myeloproliferative neoplasms (MPNs) is heterogeneous even among patients with the same MPN diagnosis. Furthermore, these clusters are not direct surrogates for prognostic scores as even patients with low and intermediate-risk scores can suffer from significant symptom burden. Recognition of symptom burden in individual patients with myelofibrosis may affect the choice, timing, and goals of therapy, as well as underscore the need for serial assessment of symptoms in a clinical setting.
MPN-SAF TSS, also known as MPN10, includes 10 items: fatigue, concentration, early satiety, inactivity, night sweats, itching, bone pain, abdominal discomfort, weight loss, and fever. The tool appears to be an efficient, sensitive, and reliable instrument for assessing symptom burden in MPN subpopulations. It has the potential to evaluate response to treatment and track disease progression.
The tool has been validated in a prospective study of 1433 patients and results correlated with other measures of disease burden. It can be used to:
- Quantitatively assess the burden of symptoms in patients with MPNs
- Track disease progression and response to treatment, facilitating disease management
- Enhance communication between physicians and patients
MPN Symptom Assessment Form Total Symptom Score
Treatment options
The goals of therapy in PV are prevention of occurrence or recurrence of thrombosis, control of hematocrit and normalization of other blood counts, and mitigation of disease-related symptoms.
Patients should be counselled that current approaches are aimed at maximizing benefits while minimizing potential risks, and are non-curative.
Goals of PV Treatment
- Control blood counts
- Prevent thromboembolic events
- Remove or reduce MPN associated symptoms
- Prevent progression to myelofibrosis or acute leukaemia
- Reduce or eliminate JAK2 mutant clone
Treatment algorithm
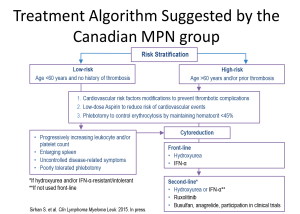
Download PDF
The European LeukemiaNet (ELN) guidelines for Philadelphia-Negative Classical Myeloproliferative neoplasms (MPNs) recommend that all patients with PV be managed with phlebotomy to maintain hematocrit below 45%, and low-dose Aspirin® to reduce risk of cardiovascular events.46
Low-risk patients
Treatment options for low risk patients include phlebotomy and low-dose Aspirin.47
Phlebotomy
Phlebotomy has been the mainstay of treatment for PV for the past 100 years.48
Although phlebotomy has the advantage of immediately reducing the hematocrit (Hct), it does not decrease the platelet or leukocyte counts due to the short half-life of these cells.
Based on several studies, the recommended Hct target is <45%.49
Although inconvenient, phlebotomies are usually well tolerated. Some patients might experience fatigue after the procedure which may be managed by adequate hydration. Frequent phlebotomies may lead to iron deficiency, abnormal red blood cell morphology, and eventually reactive thrombocytosis. Rarely, phlebotomy-induced iron deficiency might lead to complications such as increasing fatigue and restless leg syndrome.50
Low-dose Aspirin
The randomized, placebo-controlled, European Collaboration on Low-Dose Aspirin in Polycythemia Vera (ECLAP) study demonstrated a significant risk reduction in a combined endpoint of cardiovascular and venous thrombotic events (RR 0.40) with the use of low-dose Aspirin (100 mg daily) over placebo, with no increased risk of bleeding.51
Based on this data, daily low-dose Aspirin is recommended for all PV patients in the absence of contraindications.52
High-risk patients
High risk of thrombosis (>60 years of age and/or prior history of thrombosis) is the main indication for cytoreductive therapy.53
In addition, cytoreductive therapy can be considered on an individual case basis (irrespective of risk) in patients with any of the following features:
- Extreme thrombocytosis with platelet count ≥1500×109/L
- Progressive leukocytosis ≥25×109/L
- Symptomatic splenomegaly
- Severe disease-related symptoms
- Intolerance to phlebotomy, especially in patients with compromised cardiac function, inability to comply with phlebotomy requirements, or poor venous access
First line therapies
The European LeukemiaNet (ELN) guidelines recommend either hydroxyurea or interferon-α as first-line cytoreductive therapy.54
Hydroxyurea
Hydroxyurea (HU) is an oral antimetabolite that prevents DNA synthesis by inhibiting the enzyme ribonucleoside reductase.
In the Polycythemia Vera Study Group (PVSG) trial, patients treated with HU had a lower incidence of thrombosis compared with historical controls treated with phlebotomy (9.8% vs. 32.8%).55
Based on this perceived risk-benefit ratio, HU is widely used for the treatment of PV in Canada and is usually well tolerated.
The starting dose of HU is 500 mg/day, with dose increases until the desired response is obtained.
When selecting an appropriate dose of HU, the clinician should consider the extent of myeloproliferation (higher doses in cases of leukocytosis, thrombocytosis, and splenomegaly), symptom burden, and the patient’s ability to tolerate higher doses.
Female patients should be advised that HU is contraindicated in pregnancy and, therefore, appropriate contraceptive precautions should be taken.
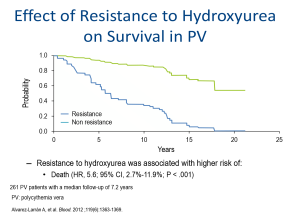
Although not a common problem in PV, the development of HU resistance or intolerance needs to be further examined, especially with the availability of other treatment options.56
In order to assist clinicians facing the decision to discontinue HU and move on to second-line therapies, an ELN panel of experts has developed a standardized definition of resistance and intolerance to HU in PV.57
Interferon
Both short-acting and pegylated interferon (IFN)-α are effective in controlling blood counts in most patients, and may be considered first-line therapy, particularly in younger patients.58
Apart from the absence of leukemogenic risk, the other benefit of IFN may be better disease control and improvement in symptoms, including pruritus.
A small French study has demonstrated that IFN reduces the size of the malignant clones measured by JAK2 allele burden and may delay or reverse fibrosis.59
IFN is commonly administered subcutaneously at a starting dose of 3 million units daily and pegylated IFN is given at a starting dose of 45-90 µg weekly. The dose should be titrated individually based on efficacy and toxicity.
Side effects, including autoimmune disorders, flu-like manifestations, depression, and heart and ocular disease lead to permanent discontinuation in 20% to 40% of patients on conventional IFN and 20% to 25% on pegylated IFN.60
Second line therapies
Other agents helpful in specific circumstances include busulfan and anagrelide.
Busulfan
Busulfan is an alkylating agent that has been demonstrated to be safe for short-term use in the elderly population, where the recognized risk of increased AML transformation with prolonged use may not be clinically relevant.61
Because of possible prolonged and delayed myelosuppression, the dose of busulfan needs to be carefully titrated.
A starting dose of 2-4 mg daily until target hematocrit level is reached is commonly used.
Anagrelide
Anagrelide disrupts the post-mitotic phase of megakaryocyte development by an unknown mechanism.62
- It is effective at lowering the platelet count without affecting other cell lineages.
- It is used only to decrease the platelet count where this causes ongoing symptoms and HU is ineffective or not tolerated.
Emerging role of ruxolitinib
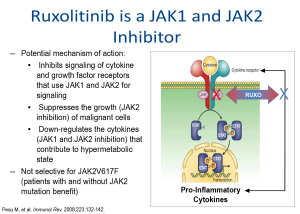
Ruxolitinib is a JAK1/JAK2 inhibitor that has demonstrated clinical benefit in patients with MPNs.63
The RESPONSE trial (NCT01243944), compared ruxolitinib to investigator-determined best available therapy (BAT). The primary endpoint (composite of Hct control and spleen volume reduction ≥35% from baseline to week 32) was achieved by 21% of patients in the ruxolitinib group vs. 1% of those in the BAT group (OR 28.64; 95% CI 4.50-1206; P <0.0001). The probability of maintaining the primary response in the ruxolitinib arm for ≥ 80 weeks from time of response was 92%.64
In the RESPONSE trial, ruxolitinib was effective in controlling disease-related symptoms and improving quality of life.
The most commonly reported non-hematologic adverse events (AEs) with ruxolitinib are headache, diarrhea, and fatigue; these are largely low-grade.
The most common hematologic AEs were anemia and thrombocytopenia; however, no patient in the RESPONSE trial discontinued treatment due to these cytopenias.
Ruxolitinib is now approved by Health Canada for treatment of PV patients resistant to or intolerant of a cytoreductive agent. A funding mechanism for ruxolitinib for PV patients has yet to be announced.
Footnotes
1 Dameshek W. Blood. 1951;6(4):372-375; Tefferi A1, Barbui T. Am J Hematol. 2015;90(2):162-173.
2 Mesa RA, et al. Cancer. 2007;109(1):68-76; Scherber R, et al. Blood. 2011;118(2):401-408; Geyer HL, et al. Blood. 2014;123(24):3803-3810.
3 Marchioli R, et al. J Clin Oncol. 2005;23(10):2224-2232.
4 James C, et al. Nature. 2005;434(7037):1144-1148; Scott LM, et al. N Engl J Med. 2007;356(5):459- 446.
5 Stuart BJ, Viera AJ. Am Fam Physician. 2004;69(9):2139-2144.
6 Marchioli R, et al. J Clin Oncol. 2005;23(10):2224-2232.
7 Passamonti F, et al. Blood. 2008;111(7):3383-3387.
8 Finazzi G, et al. Blood. 2005;105(7):2664-2670.
9 Tefferi A, et al. Leukemia. 2013S;27(9):1874-1881; Hultcrantz M, et al. J Clin Oncol. 2012;30(24):2995-3001.
10 Marchioli R, et al. J Clin Oncol. 2005;23(10):2224-2232.
11 Titmarsh GJ, et al. Am J Hematol. 2014;89(6):581-587; Mehta J, et al. Leuk Lymphoma. 2014;55(3):595-600
12 Tefferi A, et al. Leukemia. 2013;27(9):1874-1881.
13 Verstovsek S, et al. ASCO 2014. Abstract #7026.
14 Mesa RA, et al. Cancer. 2007;109(1):68-76; Scherber R, et al. Blood. 2011;118(2):401-408.
15 Siegel FP, et al. Am J Hematol. 2013;88(8):665-669; Saini K, et al. Eur J Clin Invest. 2010;40(9):828-834.
16 van Genderen PJ, Michiels JJ. Semin Thromb Hemost. 1997;23(4):357.
17 Mesa RA, et al. Cancer. 2007;109(1):68-76; Scherber R, et al. Blood. 2011;118(2):401-408.
18 Abelsson J, et al. Leuk Lymphoma. 2013;54(10):2226-2230.
19 Verstovsek S, et al. EHA 2015, Abstract #P673.
20 Thiele J, et al. In: Swerdlow S, Campo E, Harris N, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008:40-43.
21 Barbui T, et al. Leukemia. 2014;28(6):1191-1195.
22 Barbui T, et al. Am J Hematol. 2014;89(6):588-590; Barbui T, et al. Am J Hematol. 2014;89(2):199-202; Lussana F, et al. Br J Haematol. 2014;167(4):541-546.
23 Passamonti F. Blood. 2012;120(2):275-284.
24 Tefferi A. Am J Hematol. 2013;88(6):507-516
25 Mossuz P, et al. Haematologica 2004;89(10):1194-1198; Messinezy M, et al. Br J Haematol. 2002;117(1):47-53.
26 Tefferi A. Am J Hematol. 2013;88(6):507-516.
27 Tefferi A, et al. Blood. 2007;110(4):1092-1097.
28 Gangat N, et al. Eur J Haematol. 2008;80(3):197-200.
29 Andrieux JL, Demory JL. Curr Hematol Rep. 2005;4(3):224-229.
30 Swolin B, et al. Blood. 1988;72(2):386-395.
31 Tefferi A. Am J Hematol. 2013;88(6):507-516.
32 Hultcrantz M, et al. J Clin Oncol. 2012;30(24):2995-3001.
33 Marchioli R, et al. J Clin Oncol. 2005;23(10):2224-2232.
34 Landolfi R, et al. N Engl J Med. 2004;350(2):114-124; Marchioli R, et al. J Clin Oncol. 2005;23(10):2224-2232; Barbui T, et al. Blood. 2014;124(19):3021-3023; Marchioli R, et al. N Engl J Med. 2013;368(1):22-33; Kroll MH, et al. Blood Rev. 2015;29(4):215-221.
35 Tefferi A, et al. Leukemia. 2013;27(9):1874-1881; Barbui T, et al. J Clin Oncol. 2011;29(6):761-770.
36 Marchioli R, et al. J Clin Oncol. 2005;23(10):2224-2232.
37 Tefferi A, et al. Leukemia. 2013S;27(9):1874-1881.
38 Passamonti F, et al. Blood. 2008;111(7):3383-3387.
39 Passamonti F, et al. Leukemia. 2010;24(9):1574-1579; Marchioli R, et al. J Clin Oncol. 2005;23(10):2224-2232.
40 Tefferi A, et al. Leukemia. 2013S;27(9):1874-1881; Finazzi G, et al. Blood. 2005;105(7):2664-2670.
41 Kennedy JA, et al. Blood. 2013;121(14):2725-33; Passamonti F, et al. Cancer. 2005;104(5):1032-1036.
42 Anderson TJ, et al. Can J Cardiol. 2013;29(2):151-167.
43 Scherber R, et al. Blood. 2011;118(2):401-408.
44 Emanuel RM, et al. J Clin Oncol. 2012;30(33):4098-4103.
45 Tefferi A, et al. Leukemia. 2013;27(9):1874-1881.
46 Barbui T, et al. J Clin Oncol. 2011;29(6):761-770.
47 Barbui T, et al. J Clin Oncol. 2011;29(6):761-770.
48 Osler W. Lancet. 1908;1:143-146.
49 Pearson TC, Wetherley-Mein G. Lancet. 1978;2(8102):1219-1222; Landolfi R, et al. N Engl J Med. 2004;350(2):114-124; Marchioli R, et al. N Engl J Med. 2013;368(1):22-33.
50 Kim J, et al. J Nutr Biochem. 2014;25(11):1101-1107; Tobiasson M, et al. Med Oncol. 2010;27(1):105-107.
51 Landolfi R, et al. N Engl J Med. 2004;350(2):114-124.
52 Barbui T, et al. J Clin Oncol. 2011;29(6):761-770.
53 Barbui T, et al. J Clin Oncol. 2011;29(6):761-770.
54 Barbui T, et al. J Clin Oncol. 2011;29(6):761-770.
55 Fruchtman SM, et al. Semin Hematol. 1997;34(1):17-23.
56 Alvarez-Larrán A, et al. Blood. 2012;119(6):1363-1369.
57 Barosi G. et al. Br J Haematol. 2010;148(6):961-963.
58 Kiladjian JJ, et al. 2008;112(8):3065-3072.
59 Quintas-Cardama A, et al. J Clin Oncol. 2009;27(32):5418-5424; Silver RT, et al. ASH 2013 Abstract 4053.
60 Kiladjian JJ, et al. 2008;112(8):3065-3072.
61 Shvidel L, et al. Leukemia. 2007;21(9):2071-2072.
62 Anagrelide Study Group. Am J Med. 1992;92(1):69-76.
63 Verstovsek S, et al. N Engl J Med. 2012; 366(9):799-807; Harrison C, et al. N Engl J Med. 2012;366(9):787-798.
64 Vannucchi AM, et al. N Engl J Med. 2015;372(5):426-435.