Myelofibrosis
Introduction | Diagnosis | Prognostic Risk Factors | Assessment Tools | Treatment Options
Introduction
Myelofibrosis (MF) is a clonal stem cell disorder characterized by cytopenias, splenomegaly, marrow fibrosis, and systemic symptoms due to elevated inflammatory cytokines.1
About the disease
Primary myelofibrosis (PMF) is a clonal disease of hematopoietic stem cells. It is characterized by cytopenias, leukoerythroblastic blood picture, marrow fibrosis, and extramedullary hematopoiesis. A disease phenotype similar to PMF occurs in the natural history of polycythemia vera (PV) and essential thrombocythemia (ET), known as post-PV myelofibrosis (PPV-MF) and post-ET myelofibrosis (PET-MF).2
Patients with MF have significant disease burden, with debilitating symptoms that are persistent, progressive, and detrimental to quality of life.
At present, it is understood that myelofibrosis originates from acquired mutations that target the hematopoietic stem cells and induce dysregulation of kinase signaling, clonal myeloproliferation, and abnormal cytokine expression.3
Discovery of the valine-to-phenylalanine (V617F) mutation of the Janus kinase (JAK)2 gene in 60% of patients with PMF or post-ET MF and 95% of those with post-PV MF represented an important step in the understanding of the pathogenesis of MF.
The V617F mutation activates JAK2, resulting in increased phosphorylation of its substrates and leading to increased cytokine responsiveness of myeloid cells. Mutations in the myeloproliferative leukemia gene (MPL) were subsequently found in 3% to 8% of patients with PMF and post-ET MF, whereas mutations in the calreticulin gene (CALR) have been observed in half of patients with PMF and post-ET MF without JAK2 and MPL mutations. Somatic mutations in other genes, such as TET2, CBL, EZH2, DNMT3A, and ASXL1 are present in some patients with myeloproliferative neoplasms, but these can co-occur with JAK2 and MPL mutations and are found in all types of myeloid cancers.4
Patients with MF have significant disease burden, with debilitating symptoms that are persistent, progressive, and detrimental to quality of life.5
The clinical manifestations of MF are heterogeneous. Up to 30% of patients are initially asymptomatic. Most patients with MF present with anemia, splenomegaly or constitutional symptoms. During their clinical course, most patients experience massive hepatosplenomegaly along with progressive anemia requiring frequent red blood cell transfusions.6
Natural history
The clinical course of myelofibrosis (MF) is highly heterogeneous, and the disease can last from months to decades without distinct stages or clear progression.7
As the disease evolves, all patients become symptomatic due to marrow failure, increasing splenomegaly causing abdominal symptoms and early satiety, and constitutional symptoms such as weight loss, night sweats, and low-grade fever. In the advanced phases, extramedullary hematopoiesis in sites other than the spleen and liver can be seen. In addition, MF can lead to debilitating complications such as chronic thromboembolic pulmonary hypertension and portal hypertension. These may manifest at any time during the disease course. Evolution of primary MF to acute myeloid leukemia (AML) occurs at a rate of 8% to 30%.8
The 10-year survival of PMF patients is 81% lower than that of the general population.9
In a large series of 1,054 consecutive cases, the most common cause of death was transformation to acute myeloid leukemia (31% of deaths). Other causes of death included PMF progression (18%), thrombosis and cardiovascular complications (13%), infection (11%) or bleeding out of the setting of acute transformation (5%), and portal hypertension (4%).10
Patients with MPNs including those with MF have significantly more comorbidities compared with the general population. Comorbidities have a significant negative impact on the survival of these patients.11
Patients with MF are also at increased risk of infections. Multivariate analysis indicated unfavourable karyotype and JAK2 /CALR /MPL negativity as risk factors for infection.12
Epidemiology
According to the latest data, the estimated incidence of myelofibrosis (MF) is 0.1-1.0 per 100,000 worldwide. There are great variations in reported prevalence ranging from 0.5 to 2.7 per 100,000 in Europe and from 4 to 6 per 100,000 in the United States.13
The median age of MF patients at diagnosis is 69 years and < 15% are under 50 at the time of diagnosis.14
The causes of MF are largely unknown. However, case reports and a large population study suggested that genetic factors play a role in the etiology of MPNs.15
Signs and symptoms
Patients with myelofibrosis (MF) have significant debilitating symptoms, physical disabilities, and poor health-related quality of life (HRQoL).16
Symptoms of MF are caused by chronic inflammation and abnormal cytokine production, myeloproliferation and insufficient numbers of normal blood cells and splenomegaly.17
- Symptoms of chronic inflammation include night sweats, muscle and bone pain, itching (pruritus) and fever.
- Myeloproliferation leads to anemia and associated fatigue, weakness, or shortness of breath with mild exertion. Easy bleeding or bruising is a result of low platelet counts or compromised blood coagulation. Susceptibility to infections is a result of low white blood cell count.
- Fullness, discomfort, or pain in the left upper area of the abdomen and early satiety are results of an enlarged spleen pressing on the stomach and other organs. This can lead to weight loss and malnutrition. A progressively enlarging spleen can also lead to portal hypertension. This can cause varices within the stomach and esophagus, which may rupture and bleed. Liver function may be compromised as well.
The disease-related symptoms cause significant impairment in quality of life (QoL) and daily functioning. The QoL of patients with MF is similar to those with advanced solid tumors and other hematologic malignancies.18
Diagnosis
Approximately 30% of patients with myelofibrosis (MF) are asymptomatic at diagnosis and investigation is initiated after a routine physical examination reveals splenomegaly, anemia, leukocytosis, and/or thrombocytosis.

30% of MF patients are asymptomatic at diagnosis
Patients with suspected MF must undergo a careful clinical examination to rule out other possible causes of thrombocytosis or bone marrow fibrosis, including other myeloproliferative neoplasms.19
Diagnostic criteria
 The diagnosis of primary myelofibrosis (PMF) is based on well-accepted World Health Organization (WHO) 2008 criteria that include clinical, morphologic, cytogenetic and molecular assessments.20
The diagnosis of primary myelofibrosis (PMF) is based on well-accepted World Health Organization (WHO) 2008 criteria that include clinical, morphologic, cytogenetic and molecular assessments.20
Recent advances in the understanding of the pathophysiology of myelofibrosis led to the proposal to update the diagnostic criteria for PMF.21
Secondary myelofibrosis (post-polycythemia vera myelofibrosis [PPV-MF] and post-essential thrombocythemia myelofibrosis [PET-MF]) are diagnosed according to diagnostic criteria developed by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT).22
For PMF, differential diagnoses include bone marrow fibrosis associated with non-neoplastic or other neoplastic conditions such as metastatic cancer, lymphoid neoplasm, or another myeloid malignancy (i.e., chronic myeloid leukemia [CML], myelodysplastic syndromes [MDS], chronic myelomonocytic leukemia [CMML], or acute myeloid leukemia [AML]).23

Key investigations
Physical examination
Physical examination involves spleen palpation; 90% of patients with myelofibrosis have an enlarged spleen. Hepatomegaly is present in about 50% of patients.24
Complete blood count (CBC) and peripheral blood smear
A CBC is usually obtained to evaluate the hemoglobin level, white blood cell count and platelet count. The peripheral smear of patients with primary myelofibrosis includes red blood cells of variable shape (including tear drop cells) and size and variable degrees of polychromasia. Leukoerythroblasosis is present in most cases and is recognized by the presence of immature cells from the myeloid and erythoblasic lineages.25
Biochemistry
Patients with MPNs may have nonspecific abnormalities in a variety of biochemistry tests. These include: increased lactate dehydrogenase (LDH), uric acid, and vitamin B12. It is noteworthy that increased LDH is not specific for PMF, and can also be seen in polycythemia vera (PV) and essential thrombocythemia (ET). An increase in LDH may be due to ineffective hematopoiesis or hemolytic process occurring in the spleen. Hyperuricemia is due to enhanced turnover of hematopoietic tissue.26
Mutation testing
Testing for the JAK2 V617F mutation should be performed early during the diagnostic workup of suspected MPNs. JAK2 V617F negative patients who meet other criteria for ET or PMF should be screened for CALR mutation. MPL mutation testing should be reserved for patients who are negative for JAK2 V617F and CALR mutations and in whom bone marrow biopsy and other investigations support diagnosis of PMF.27
Bone marrow evaluation
Evaluation of bone marrow histopathology is critical for correct diagnosis of MF. The bone marrow biopsy of a patient with MF typically reveals megakaryocyte proliferation and atypia, usually with reticulin or collagen fibrosis.28
A bone marrow (BM) biopsy report should include: age adjusted cellularity and presence of fibrosis (reticulin and collagen stains), evaluation of granulopoiesis with special reference to blast clusters, and evaluation of erythropoiesis (frequency and distribution).29
Occasionally, overt bone marrow fibrosis might be absent (i.e., prefibrotic PMF) and, in the presence of thrombocytosis, a patient may be misdiagnosed with essential thrombocythemia (ET). The possibility of prefibrotic primary myelofibrosis, as opposed to ET, should be considered in the presence of persistently increased serum LDH, anemia, leukoerythroblastosis, increased circulating CD34+ cell count, and splenomegaly. The distinction between ET and prefibrotic PMF is clinically relevant because both overall and leukemia-free survival are significantly worse in prefibrotic PMF.30
Supportive investigations
Cytogenetics
In patients with myelofibrosis (MF) cytogenetic abnormalities carry significant prognostic value and are included in the Dynamic International Prognostic Scoring System (DIPSS) Plus.31
Approximately one-third of patients with primary MF present with cytogenetic abnormalities. The most frequent are del(20q), del(13q), trisomy 8 and 9, and abnormalities of chromosome 1 including duplication 1q. Other less frequent lesions include -7 ⁄ del(7q), del(5q), del(12p), +21 and der(6)t(1;6)(q21;p21.3). Unfavourable karyotypes are associated with thrombocytopenia, leukopenia, circulating blasts ≥1%, and lower hemoglobin levels. Available data also suggest that patients with unfavourable karyotypes are at high risk of leukemic transformation.32
Recent studies have identified that mutations in several genes (ASXL1, EZH2, SRSF2 and IDH1/2) have a significant impact on disease progression and survival in MF. Harbouring more than one mutation in any of these genes is considered a “high molecular risk” and is associated with reduced survival and increased risk of blast transformation in PMF.33
Thus, it has been proposed to incorporate these mutations in prognostic scoring systems. Currently, however, testing for these genes is done for research purposes only and not in routine clinical practice.
Download PDF
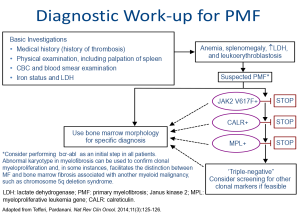
Diagnostic algorithm for MF
The diagnostic work-up for suspected primary myelofibrosis (PMF) should start with medical history and physical examination, followed by complete blood counts and blood smear examination. PMF is suspected in cases of anemia, splenomegaly, increased lactate dehydrogenase, and leukoerythroblastosis. These patients should undergo mutation testing and bone marrow investigations.
Prognostic risk factors
Treatment of myelofibrosis (MF) is largely risk- and problem-oriented and individualized for each patient. To support this individualization of treatment, several risk stratification systems have been developed. The International Prognostic Scoring System (IPSS) estimates prognostic risk at the time of diagnosis, whereas the Dynamic IPSS (DIPSS) is used for re-evaluating survival predictions during the course of disease progression and treatment.34 DIPSS Plus further refines DIPSS by incorportating information from karyotype, platelet count, and transfusion status35.
The prognosis of MF is different for every patient. People in a good prognostic group can live for many years without having major symptoms. Those with a poor prognosis may progress more quickly.
International Prognostic Scoring System (IPSS)
The IPSS is considered the gold standard for risk assessment in patients newly diagnosed with primary myelofibrosis (PMF).
The IPSS was developed by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) using a multi-institutional series of more than 1,000 patients with primary myelofibrosis (PMF). The IPSS includes 5 variables, measured at diagnosis, for prediction of survival: age older than 65 years, the presence of constitutional symptoms, hemoglobin level < 100 g/L, leukocyte count > 25 × 109/L, and 1% or more blasts in the peripheral blood.36
An IPSS calculator is available online in the MPN eSIMPLE app.
Dynamic International Prognostic Scoring System (DIPSS)
DIPSS includes additional risk factors not present at diagnosis, the acquisition of anemia in particular. It is a useful tool to monitor patients during the course of their disease.37
DIPSS Plus was developed using 793 consecutive patients from the Mayo Clinic database. The additional prognostic variables included in the DIPSS Plus include red blood cell transfusion need, thrombocytopenia, and unfavourable karyotype. Each variable is assigned one point.38
A DIPSS calculator is available online in the MPN eSIMPLE app.
DIPSS Plus
The DIPSS Plus risk stratification system refines DIPSS by incorporating prognostic information from karyotype, platelet count, and transfusion status.
The Dynamic International Prognostic Scoring System (DIPSS) Plus was developed using 793 consecutive patients from the Mayo Clinic database. The additional prognostic variables included in the DIPSS Plus include red blood cell transfusion need, thrombocytopenia, and unfavorable karyotype. Each variable is assigned one point.39
A DIPSS tool is available online in the MPN eSIMPLE app.
Tools under investigation
Emerging research has identified alternative approaches to assess prognostic risk in MF based on the presence or absence of specific gene mutations (i.e., ASXL1, EZH2, SRSF2, IDH1, IDH2, CALR, and MPL). Ongoing investigations are focused on the development of integrated systems for evaluating prognostic risk in patients with MF using molecular and cytogenetic markers in combination with clinical and hematologic findings. Two new prognostic scoring systems, both modifications of the IPSS, have been proposed.40
Mutation-enhanced International Prognostic Scoring System (MIPSS)
MIPSS is a collaborative project between Italian and international researchers (Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative [AGIMM]; International Working Group for Myelofibrosis Research and (IWG-MRT].41
- In a multivariable analysis, age > 60 years, constitutional symptoms, hemoglobin <100g/L, platelets <200×109/L, triple negativity (JAK2, MPL and CALR), presence of JAK2 or MPL mutations, or ASXL1 and SRSF2 mutations were significantly associated with reduced survival. Based on this, four distinct risk groups were identified.
- The proposed MIPSS addresses the mutation-relevant prognostic information gap in IPSS and performs equally well in the setting of patients seen at time of diagnosis or over the disease course.
- According to the MIPSS up to 67% of patients in the intermediate I category of the IPSS were upstaged to an intermediate II or high-risk MIPSS. The MIPSS also downstaged 50% of IPSS high-risk patients.
Genetics-based Prognostic Scoring System (GPSS)
An alternative genetics-based prognostic scoring system (GPSS) complements the MIPSS. In addition to high-risk mutations and age, the GPSS includes high-risk karyotypes: 5q-, +8, inv(3), i(17q), -7/7q-, 11q or 12p abnormalities, autosomal trisomies except +9, monosomal and complex non-monosomal karyotypes.42
Symptom assessment
Myelofibrosis (MF) not only shortens survival but also severely compromises quality of life (QoL).
Compromised QoL is a result of marked splenomegaly and profound constitutional symptoms that are believed to be cytokine-mediated. MF-related symptoms restrict participation in both social functions and physical activities. In addition, a third of myeloproliferative neoplasms (MPNs) patients need assistance with activities of daily living. As expected, the presence of myelofibrosis, anemia, splenomegaly, or other features associated with advanced disease lead to a higher degree of fatigue. In fact, MPN patients, including those with MF, suffer from substantial fatigue that is in excess of what is expected from age-matched controls and not necessarily attributed to the presence of anemia.43
Myelofibrosis-related symptoms are troublesome to patients, and alleviation of this burden is a paramount treatment objective.44
Assessment Tools
Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF TSS)
A recent cluster analysis suggested that symptom burden in myeloproliferative neoplasms (MPNs) is heterogeneous even among patients with the same MPN diagnosis. Furthermore, these clusters are not direct surrogates for prognostic scores as even patients with low and intermediate-risk scores can suffer from significant symptom burden. Recognition of symptom burden in individual patients with myelofibrosis may affect the choice, timing, and goals of therapy, as well as underscore the need for serial assessment of symptoms in a clinical setting.45
MPN-SAF TSS, also known as MPN10, includes 10 items: fatigue, concentration, early satiety, inactivity, night sweats, itching, bone pain, abdominal discomfort, weight loss, and fever. The tool appears to be an efficient, sensitive, and reliable instrument for assessing symptom burden in MPN subpopulations. It has the potential to evaluate response to treatment and track disease progression.
The tool has been validated in a prospective study of 1433 patients and results correlated with other measures of disease burden. It can be used to:
- Quantitatively assess the burden of symptoms in patients with MPNs
- Track disease progression and response to treatment, facilitating disease management
- Enhance communication between physicians and patients46
MPN Symptom Assessment Form Total Symptom Score
Treatment options
The discovery of the Janus kinase (JAK2) mutation triggered the development of targeted therapy for myelofibrosis (MF), resulting in approval of the JAK1/2 inhibitor, ruxolitinib (Jakavi®).
Although ruxolitinib changed the therapeutic approaches for MF, allogeneic hematopoietic stem cell transplant (HSCT) remains the only curative therapy. However, due to the associated morbidity and mortality, HSCT is usually restricted to eligible high- and intermediate-2-risk MF patients.
Ruxolitinib is the first MF therapy approved by Health Canada.
Eligible patients are, in general, relatively younger with fewer comorbidities. Aiming for the best symptom control possible and improvement in quality of life should be a therapeutic goal for all patients who are not fit for transplantation. Thus, it is especially important to establish individualized therapeutic goals upfront for each patient. The goals should also be revisited on a regular basis.47
Transplant
Allogeneic hematopoietic stem cell transplantation (HSCT) remains the only curative approach for myelofibrosis (MF). However, HSCT carries a considerable risk of mortality and morbidity. Significant regimen-related toxicities, graft failure and graft-versus-host disease are major barriers to the success of HSCT in MF. Use of reduced-intensity conditioning has helped to expand the applicability of HSCT to older MF patients. Age, risk score, and donor selection have a significant impact on outcomes after HSCT with reduced-intensity conditioning.48
Recent studies suggest that the Dynamic International Prognostic Scoring System (DIPSS) score may predict success after transplant.49
The DIPSS can also be used to select appropriate candidates for transplant. Patients with MF who are age ≤65 years, and have intermediate-2 or high-risk disease by DIPSS have superior survival with HSCT compared with non-transplant approaches.50
It has been demonstrated that pretreatment with JAK1/2 inhibitors has no adverse impact on the outcomes of subsequent HSCT. Furthermore, serious adverse events during drug discontinuation appear to be less frequent when JAK inhibitors are continued until close to the conditioning regimen. Patients undergoing HSCT while responding to JAK inhibitors appear to have better outcomes compared to those with progressive disease.51
Conventional therapies
Due to the lack of well-designed prospective studies and limited treatment efficacy consensus recommendations regarding the role of conventional agents in the management of myelofibrosis (MF) cannot be developed. Furthermore, none of the conventional agents has been shown to modify the natural history of the disease. Traditionally conventional therapies are used to treat the two most prominent symptoms of myelofibrosis: splenomegaly and anemia.52
Therapies for splenomegaly
Splenectomy
Traditionally, splenectomy has been used to manage burdensome symptoms associated with splenomegaly, but the procedure involves substantial risk. The main complications are bleeding, infections, and thrombosis (primarily in the splanchnic veins). The decision to undertake splenectomy should be made carefully, balancing the risks against the possible benefits.53
Splenic irradiation
Splenic irradiation has been used in selected patients for palliative purposes when splenomegaly is resistant to medication and a splenectomy is contraindicated due to advanced age or significant co-morbidities. The doses used range from 30-365 Gy in 5-10 fractions. Toxicities include severe cytopenias (seen in about 12-35% patients) and an increase in transfusion requirements (seen in approximately 40% of cases).54
Cytoreductive therapies
Hydroxyurea was for many years the therapy of choice for MF splenomegaly. Presently, hydroxyurea can be an option for moderately symptomatic splenomegaly, especially in patients not eligible for JAK inhibitors. Other drugs such as busulfan or melphalan are rarely used.55
Treatment of MF-related anemia
Conventional treatment options include androgens, erythropoietic stimulating agents (ESAs) or immunomodulators either alone or in combination with prednisone.56
Androgens
The androgens oxymetholone and danazol are widely used in clinical practice despite the fact that only a few small studies have examined their efficacy in MF.57
Erythropoietin
Erythropoietin (EPO) is a reasonable treatment option for selected MF patients with anemia and low EPO levels. Careful monitoring of spleen size is required as the use of EPO can result in splenomegaly.58
Immunomodulators
Immunomodulators include thalidomide and the next-generation immunomodulatory drugs (IMiDs) lenalidomide and pomalidomide. Poor tolerability and significant side effects (mainly hematologic toxicity) are major concerns with these drugs in MF and preclude their wider use. To reduce toxicities, low-dose prednisone can be used.59
Ruxolitinib
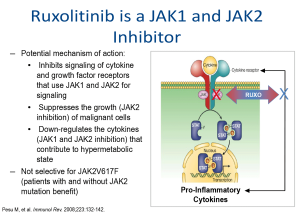
Ruxolitinib is a JAK1/JAK2 inhibitor that suppresses cytokine and growth factor receptors that use JAK1 and JAK2 for signalling.60
Regulatory approval in Canada was based on the results of two pivotal randomized phase III trials: Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment (COMFORT)-I and COMFORT II. In both COMFORT- I and -II, treatment with ruxolitinib led to significant reduction in spleen size and improvement is symptom burden and quality of life. There was no significant difference in response among patients with or without the JAK2 V617F mutation.61
A small phase I/II trial demonstrated that ruxolitinib also improves exercise capacity and body weight.62
Longer follow-up confirmed a survival advantage for ruxolitinib treatment.63
The COMFORT trials also demonstrated significant reduction in allele burden that continues over time and is correlated with reductions in spleen volume.64
Overall, ruxolitinib is well tolerated, with the main toxicity being hematological. In COMFORT I, Grade 3-4 hematological effects occurring more frequently with ruxolitinib included anemia (45.2% vs. 19.2%), thrombocytopenia (12.9% vs. 1.3%), and neutropenia (7.1% vs. 2.0%). However, the platelet count and hemoglobin levels tend to stabilize and improve over time and do not affect response to ruxolitinib treatment.65
The recommended starting dose of ruxolitinib is based on platelet count. For a platelet count >200 x 109/L, the recommended starting dose is 20 mg BID and for a platelet count 100-200 x 109/L, the recommended dose is 15 mg twice daily (BID). A dose of 15 mg BID may also be considered in transfusion independent patients, who may have difficulty tolerating a drop in hemoglobin of 20 g/L. Complete blood and platelet counts must be performed before initiating therapy, every 2 to 4 weeks until doses are stabilized, and then as clinically indicated. Dose reduction should be considered for patients receiving ruxolitinib 15 or 20 mg BID if the platelet count declines below 100 x 109/L. When treatment interruption is required, dose tapering is advised. Dose increases in increments of 5 mg BID can be considered on a monthly basis to a maximum dose of 25 mg BID in patients with inadequate response if no significant hematological toxicity occurs.66
Preliminary data from EXPAND (a phase Ib, dose-finding study in patients with low platelet counts) indicates that ruxolitinib starting doses of 10 mg and 15 mg BID are well tolerated in patients with baseline platelet counts of 50-74 × 109/L and 75-99 × 109/L, respectively, and provide meaningful improvement in disease-related symptoms.67
Clinical trials/Emerging therapies
Several other JAK inhibitors are in various stages of clinical development: pacritinib and momelotinib are the most advanced.
In a phase I/II study of momelotinib in myelofibrosis (MF) the rates of spleen and anemia response, per International Working Group criteria, were 48% and 59%, respectively.68
Momelotinib is currently being evaluated in two phase III clinical trials in JAK inhibitor naïve patients (SIMPLIFY 1; NCT01969838) and those previously treated with JAK inhibitors (SIMPLIFY 2; NCT02101268)
PERSIST-1, a randomized phase III trial, assessed the efficacy and safety of pacritinib in patients with primary and secondary MF vs. best available therapy (BAT), excluding ruxolitinib. The trial included patients irrespective of their baseline platelet or hemoglobin levels.
- Pacratinib was effective in reducing spleen volume and improving symptoms regardless of the baseline platelet level.
- Patients with baseline platelets <50 x 109/L had a 35% mean increase in platelet counts by week 24.
- In red blood cell (RBC) transfusion-dependent patients, 25.7% of patients in the pacritinib arm became RBC independent compared with 0% of those receiving BAT (P=0.043).
- The most common adverse events (AEs) with pacritinib were diarrhea (grade 3, 5%), nausea (grade 3, 0.9%), and vomiting (grade 3, 0.9%). Hematologic AEs were similar between the pacritinib and BAT arms. The symptoms typically lasted less than 1 week and few patients discontinued treatment due to side effects.69
- Pacritinib is also being evaluated in patients with MF and low platelets counts (PERSIST-2; NCT02055781).
Several ongoing trials are investigating ruxolitinib in combination with therapies that target different pathways.70
Notes
1 Abdel-Wahab OI, Levine RL.Annu Rev Med. 2009;60:233-245; Gupta V, et al. Am J Blood Res 2012;2(3):170-186.
2 Abdel-Wahab OI, Levine RL.Annu Rev Med. 2009;60:233-245; Gupta V, et al. Am J Blood Res 2012;2(3):170-186
3 Tefferi A. Blood 2011;117(13):3494-3504.
4 Tefferi A. Blood 2011;117(13):3494-3504; Klampfl T, et al. N Engl J Med. 2013;369(25):2379-2390; Cervantes F. Blood 2014;124(17):2635-2642.
5 Scherber R, et al. Blood. 2011;118(2):401-408.
6 Tefferi A et al. Semin Oncol. 1995;22(4):327; Cervantes F. et al. Blood. 2009;113(13): 2895-2901.
7 Cervantes F, et al. Blood. 2009;113(13):2895-2901; Cervantes F, et. al. Semin Oncol. 2005;32(4):395-402.
8 Cervantes F. Blood 2014;124(17):2635-2642; Gupta V, et al. Am J Blood Res 2012;2(3):170-186; Passamonti F, et al. Oncotarget. 2011;2(6):485-490.
9 Hultcrantz M, et al. J Clin Oncol. 2012;30(24):2995-3001.
10 Cervantes F, et al. Blood. 2009;113(13):2895-2901.
11 Bak M, et al. EHA 2015, Abstract P306; Newberry KJ, Cancer. 2014;120(19):2996-3002.
12 Polverelli, et al. EHA 2015. Abstract P667.
13 Mehta J, et al. Leuk Lymphoma. 2014;55(3):595-600 ; Moulard O, et al. Eur J Haematol. 2014;92(4):289-297.
14 Landgren O, et al. Blood 2008; 112 (6): 2199-2204; Cervantes F, et al. Blood. 2009;113(13):2895-2901.
15 Landgren O, et al. Blood 2008; 112 (6): 2199-2204.
16 Scherber R, et al. Blood. 2011;118(2):401-408.
17 Scherber R, et al. Blood 2011;118(2):401-408; Geyer HL, et al. Blood. 2014;123(24):3803-3810; Mesa RA, et al. Cancer 2007; 109: 68-76.
18 Cervantes F, et al. Blood. 2009;113(13):2895-2901; Scherber R, et al. Blood 2011;118(2):401-408; Geyer HL, et al. Blood. 2014;123(24):3803-3810; Mesa RA, et al. Cancer 2007; 109: 68-76; Kiladjian J-J,et al. J Clin Oncol 2012; 30: 6626.
19 Swerdlow SH, et al. Vol 2. 4th ed. Geneva: World Health Organization; 2008.
20 Thiele J, et al. IARC Press: Lyon, France, 2008, pp 40–43; Vardiman JW, et al. Blood 2009; 114: 937-951.
21 Tefferi A, et al. Leukemia. 2014;28(7):1407-1413.
22 Barosi G, et al. Leukemia. 2008;22(2):437-438.
23 Tefferi A et al. Blood. 2011;117(13):3494-3504.
24 Cervantes F, et al. Blood 2009; 113: 2895-2901.
25 Thiele J, et al. IARC Press: Lyon, France, 2008, pp 40–43; Mascarenhas JO, et al. Haematologica. 2013;98(10):1499-1509.
26 Thiele J, et al. IARC Press: Lyon, France, 2008, pp 40-43.
27 Tefferi A, Pardanani A. Nat Rev Clin Oncol. 2014;11(3):125-126.
28 Thiele J, et al. IARC Press: Lyon, France: 2008;40-43
29 Thiele J, et al. Haematologica 2005;90(8):1128-1132.
30 Thiele J, et al. Best Pract Res Clin Haematol. 2006;19(3):413-437; Barbui T, et al. J Clin Oncol. 2011;29(23):3179-3184.
31 Gangat N, et al. J Clin Oncol. 2011;29(4):392-397.
32 Tefferi A, et al. Blood. 2011;118(17):4595-4598; Caramazza D, et al. Leukemia. 2011;25(1):82-88; Hussein K, et al. Blood. 2010;115(3):496-499; Tefferi A, et al.. ASH 2014. Abstract # 406; Tefferi A, et al. ASH 2014. Abstract # 631.
33 Nangalia J, et al. N Engl J Med. 2013;369(25):2391-2405; Guglielmelli P, et al. Leukemia. 2014 28(9):1804-1810; Vannucchi AM, et al. Leukemia. 2013;27(9):1861-1869.
34 Cervantes F, et al. Blood. 2009;113(13):2895-2901; Passamonti F, et al. Blood. 2010;115(9):1703-1708; Gangat N, et al. J Clin Oncol. 2011;29(4):392-397.
35 Gangat N, et al. J Clin Oncol. 2011;29(4):392-397
36 Cervantes F, et al. Blood. 2009;113(13):2895-2901.
37 Passamonti F, et al. Blood.2010;115(9):1703-1708.
38 Gangat N, et al. J Clin Oncol. 2011;29(4):392-397.
39 Gangat N, et al. J Clin Oncol. 2011;29(4):392-397
40 Vannucchi AM, et al. ASH. 2014, Abstract 405; Tefferi A, et al. ASH 2014. Abstract 406; Tefferi A, et al. ASH 2014. Abstract 631.
41 Vannucchi AM, et al. ASH. 2014, Abstract 405.
42 Tefferi A, et al. ASH 2014. Abstract 406; Tefferi A, et al. ASH 2014. Abstract 631.
43 Mesa RA, et al. 2007 1;109(1):68-76; Messa RA, et al. Cancer 2011;117 :4869-4877.
44 Messa RA, et al. Cancer 2011;117 :4869-4877.
45 Geyer HL, et al. Blood. 2014;123(24):3803-3810.
46 Emanuel RM, et al. J Clin Oncol. 2012;30(33):4098-103.
47 Gupta V, et al. Am J Blood Res. 2012;2(3):170-186.
48 Gupta V, et al. Blood 2012; 120:1367-1379; Alchalby H et al. Br J Haematol. 2012;157(1):75-85; Kröger N, et al. Blood. 2009;114(26):5264-5270.
49 Scott BL, et al. Blood 2012; 119: 2657-2664.
50 Kröger N, et al. ASH 2014, Abstract 633.
51 Shanavas, et al. EHA 2015. Abstract S450.
52 Gupta V, et al. Am J Blood Res. 2012;2(3):170-186.
53 Cervantes F. Blood. 2014;124(17):2635-2642; Gupta V, et al. Am J Blood Res. 2012;2(3):170-186.
54 Gupta V, et al. Am J Blood Res. 2012;2(3):170-186; Elliott MA, et al. Br J Haematol 1998; 103: 505-511.
55 Gupta V, et al. Am J Blood Res. 2012;2(3):170-186; Cervantes F. Blood. 2014;124(17):2635-2642.
56 Gupta V, et al. Am J Blood Res. 2012;2(3):170-186; Cervantes F. Blood. 2014;124(17):2635-2642.
57 Gupta V, et al. Am J Blood Res. 2012;2(3):170-186.
58 Gupta V, et al. Am J Blood Res. 2012;2(3):170-186.
59 Gupta V, et al. Am J Blood Res. 2012;2(3):170-186; Cervantes F. Blood. 2014;124(17):2635-2642.
60 Pesu M, et al. Immunol Rev. 2008;223:132-142.
61 Verstovsek S, et al. N Engl J Med. 2012;366(9):799-807; Harrison CN, et al. N Engl J Med. 2012;366(9):787-798.
62 Verstovsek S, et al. N Engl J Med. 2010;363(12):1117-1127.
63 Verstovsek S, et al. Blood. 2013;122: Abstract 396; Harrison C, et al. EHA 2014, Poster P403; Vannucchi AM, et al. Blood. 2013; 122 (21): Abstract 2820.
64 Vannucchi AM, et al. ASH 2012:Abstract 802; Deininger M, et al. EHA 2015. Abstract P674.
65 Verstovsek S, et al. N Engl J Med. 2012;366(9):799-807; Verstovsek S, et al. Blood. 2013;122: Abstract 396; Verstovsek S, et al. ASCO 2011. Abstract 6500.
66 JAKAVI Product Monograph, June 15, 2012; Gupta V, et al. Am J Blood Res. 2012;2(3):170-186.
67 te Boekhorst P, et al, ASH 2014, Abstract 1841.
68 Pardanani A, et al. Leukemia 2013;2(6) 7:1322-1327.
69 Mesa RA, et al. J Clin Oncol. 33, 2015 (suppl; abstr LBA7006; Harrison C, et al. EHA 2015, Abstract LB314.
70 Durrant S, et al. ASH 2014.Abstract # 710; Kiladjian JJ, et al. ASH 2014. Abstract # 711; Gupta V, et al. ASH 2014. Abstract # 712; Verstovsek S, et al. ASH 2014. Abstract # 713.